De novo mass spectrometry peptide sequencing with a transformer model
{kind=link}
Abstract
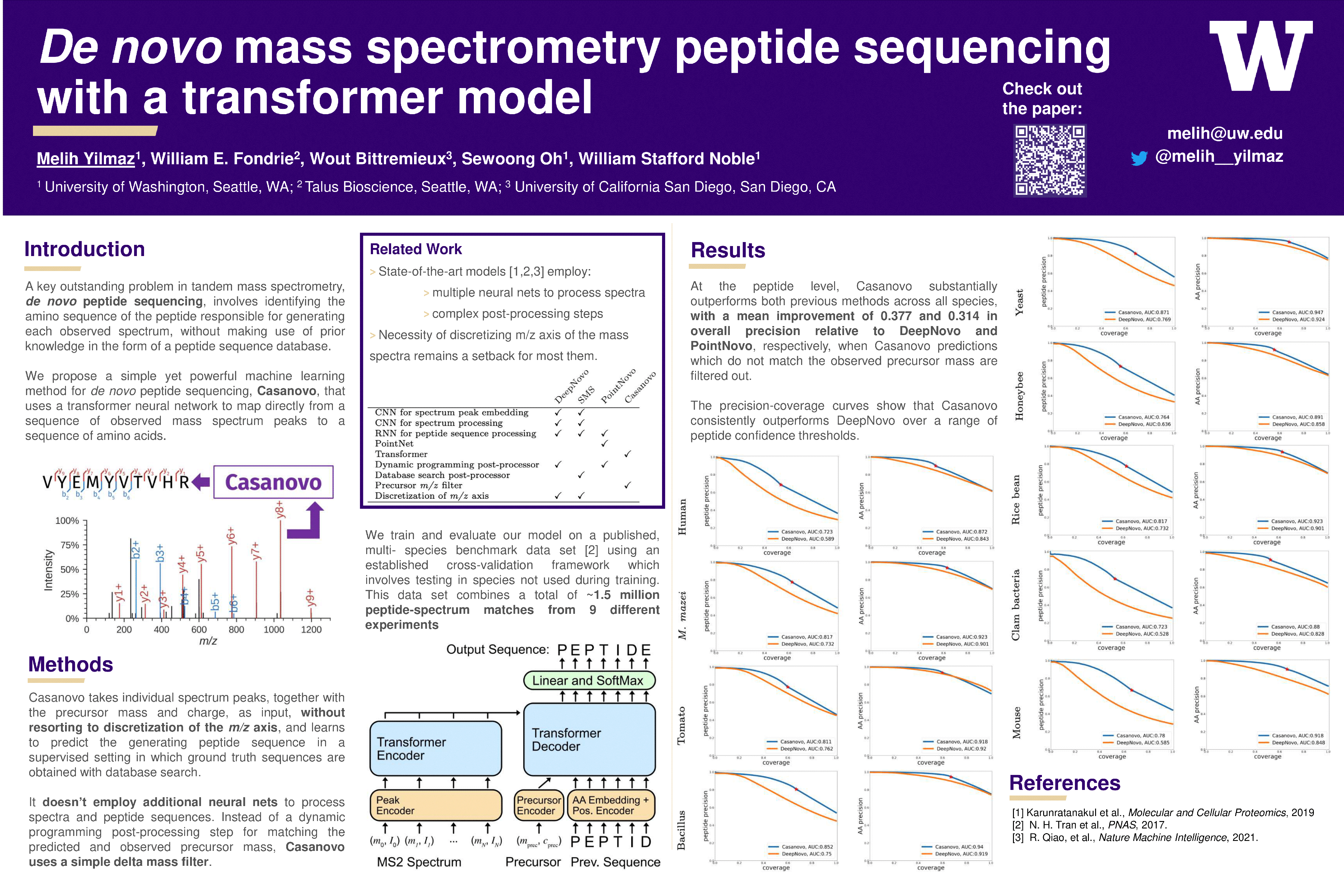
Tandem mass spectrometry is the only high-throughput method for analyzing the protein content of complex biological samples and is thus the primary technology driving the growth of the field of proteomics. A key outstanding challenge in this field involves identifying the sequence of amino acids -the peptide- responsible for generating each observed spectrum, without making use of prior knowledge in the form of a peptide sequence database. Although various machine learning methods have been developed to address this de novo sequencing problem, challenges that arise when modeling tandem mass spectra have led to complex models that combine multiple neural networks and post-processing steps. We propose a simple yet powerful method for de novo peptide sequencing, Casanovo, that uses a transformer framework to map directly from a sequence of observed peaks (a mass spectrum) to a sequence of amino acids (a peptide). Our experiments show that Casanovo achieves state-of-the-art performance on a benchmark dataset using a standard cross-species evaluation framework which involves testing with spectra with never-before-seen peptide labels. Casanovo not only achieves superior performance but does so at a fraction of the model complexity and inference time required by other methods.